Prescription required. We facilitate access to medicines already prescribed by your treating physician. We do not provide medical advice or diagnoses.
AKT Mutation Alterations in Oncology: Molecular Drivers and Targeted Inhibitor Protocols
Educational Overview
Receiving an advanced cancer diagnosis immediately necessitates a transition from generalized anatomical staging to precise molecular profiling. In modern oncology, the standard of care dictates that malignancies are treated based on their specific genetic drivers. Among the most critical discoveries in tumor genomics is the hyperactivation of the AKT signaling network.
If comprehensive genomic profiling (CGP) or a liquid biopsy indicates an AKT mutation, amplification, or a related pathway alteration (such as PTEN loss), your oncology team has identified the specific biological engine driving the malignancy. This genomic data is highly actionable. This guide details the molecular biology of the AKT kinase, the mechanism of highly selective AKT inhibitors, and the clinical implications for patients navigating advanced, endocrine-resistant disease.
The Molecular Biology of AKT (Protein Kinase B)
Inside human cellular architecture, DNA provides the exact transcriptional blueprints for protein synthesis. The AKT gene (also classified as Protein Kinase B or PKB) provides instructions for a serine/threonine-specific protein kinase that serves as the central communication node within the cell.
In a healthy physiological state, the three isoforms of the AKT protein (AKT1, AKT2, and AKT3) function as a highly regulated molecular switch governing cell survival, proliferation, and metabolic processes.
- The Regulated State: AKT is temporarily activated in response to extracellular growth signals to facilitate tissue repair. Once the physiological requirement is met, the kinase is deactivated, allowing the cell to eventually undergo apoptosis (programmed cell death). This ensures tissue homeostasis.
- The Pathological State: In malignant cells, genomic alterations cause this kinase to remain constitutively active. The AKT network is forced into a permanent, self-sustaining state of activation. The cell receives uninterrupted commands to consume systemic glucose, synthesize proteins, and aggressively evade apoptotic signals. These immortalized cells rapidly accumulate, driving tumor progression and systemic metastasis.
The Science of the Mutation: The AKT1 E17K Hotspot
To understand this pathological drive, oncologists examine the genetic code at the molecular level. While the AKT family consists of three distinct genes, the most clinically actionable somatic mutations in solid tumors occur within the AKT1 gene.
Mutations frequently localize to specific, highly vulnerable regions of the genome known as “hotspots.” In the AKT1 gene, the hallmark activating mutation is the E17K alteration.
Mechanism of the E17K Alteration
- The Amino Acid Substitution: At codon 17 of the AKT1 protein, a specific transcriptional error causes the negatively charged amino acid glutamic acid (E) to be erroneously replaced by the positively charged amino acid lysine (K).
- Electrostatic Anchoring: Proteins rely on precise electrostatic charges to fold and function. This single amino acid substitution occurs in the Pleckstrin Homology (PH) domain of the protein. The introduction of an abnormal positive charge acts as an electrostatic magnet, permanently anchoring the mutant AKT1 protein to the inner plasma membrane.
- Constitutive Activation: Normal AKT requires the presence of a specific lipid (PIP3) to localize to the membrane and activate. The E17K mutation completely bypasses this biological requirement. The kinase remains continuously active and firing, entirely independent of upstream cellular growth signals.
Clinical Prevalence Across Solid Tumors
Comprehensive molecular profiling has identified AKT pathway alterations across several malignancies, frequently acting as a primary mechanism of resistance to standard therapies:
- Breast Cancer: AKT1 E17K mutations are identified in Hormone Receptor-positive (HR+), HER2-negative breast cancers. They appear in approximately 4% to 8% of these advanced cases, heavily correlating with resistance to standard endocrine therapy.
- Gynecologic Cancers: Amplifications and point mutations within the broader PI3K/AKT pathway are frequently identified in endometrial and ovarian carcinomas, making them prime targets for emerging kinase inhibitors.
- Prostate Cancer: While isolated AKT point mutations are less common, broader PI3K/AKT pathway hyperactivation is a major biological driver in metastatic castration-resistant prostate cancer (mCRPC).
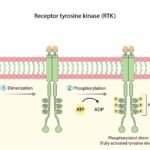
The PI3K/AKT/mTOR Signaling Cascade
AKT does not operate in a vacuum; it is the central hub of a highly interconnected cellular highway known as the PI3K/AKT/mTOR pathway. Understanding this cascade is critical to understanding how targeted therapies arrest tumor growth.
- Upstream Activation (PI3K): Receptor Tyrosine Kinases (RTKs) on the cell surface receive growth signals from the body and activate Phosphoinositide 3-kinase (PI3K). PI3K then converts a cellular lipid called PIP2 into PIP3.
- The Central Hub (AKT): PIP3 recruits the AKT protein to the cell membrane, where it is phosphorylated and fully activated. (In the presence of an E17K mutation, AKT skips the PIP3 requirement and activates itself).
- Downstream Effectors (mTOR): Once active, AKT phosphorylates multiple downstream targets, most notably activating the mTORC1 complex. mTOR functions as the cellular factory, driving rapid protein synthesis, nutrient uptake, and tumor expansion.
- The Tumor Suppressor (PTEN): PTEN is a lipid phosphatase that acts as the primary physiological brake for this pathway. It converts PIP3 back to PIP2, effectively shutting off AKT. In many advanced cancers, the PTEN gene is deleted or mutated, removing the brakes and allowing AKT to run unchecked.

When the mutated AKT protein fires continuously—or when PTEN is lost—it floods the downstream mTOR factory with hyperactive signals, resulting in aggressive tumor expansion.
Targeted Therapy: Mechanism of AKT Inhibitors
Historically, advanced solid tumors were treated with broad-spectrum, cytotoxic chemotherapy, which attacks all rapidly dividing cells and induces severe systemic toxicities. To directly combat AKT-driven tumors, pharmaceutical research has developed highly specific, small-molecule pan-AKT inhibitors (such as capivasertib).
ATP-Competitive Inhibition
AKT requires adenosine triphosphate (ATP) as cellular fuel to phosphorylate its downstream targets and sustain the tumor. Small-molecule AKT inhibitors are designed as ATP-competitive agents.
After entering the patient’s bloodstream and penetrating the cancer cell membrane, the drug physically binds to the ATP-binding cleft of all three AKT isoforms (AKT1, AKT2, and AKT3). By occupying this precise molecular pocket, the drug displaces the ATP fuel, instantly stalling the kinase’s catalytic activity.
By silencing the AKT hub, the drug effectively halts downstream mTOR signaling. The cancer cell, abruptly deprived of its continuous survival commands, ceases division and initiates programmed cell death (apoptosis).
Target Specificity and On-Target Adverse Events
Target specificity defines a drug’s ability to selectively attack cancer cells while minimizing collateral damage to healthy tissue. While AKT inhibitors largely spare unrelated healthy organ systems, the AKT2 isoform plays an essential role in normal human insulin signaling and glucose homeostasis.
When you consume carbohydrates, insulin instructs normal cells to activate AKT2, which pulls glucose out of the bloodstream for energy. Because a pan-AKT inhibitor blocks AKT universally throughout the body, it temporarily induces peripheral insulin resistance.
As a direct, on-target physiological effect, hyperglycemia (elevated blood glucose) is a primary clinical side effect of this drug class. Oncologists proactively manage this through baseline glucose monitoring, dietary modifications, and the concurrent administration of standard anti-diabetic agents (such as metformin) to maintain metabolic stability without interrupting the oncology treatment. Other common, manageable side effects include grade 1/2 diarrhea and cutaneous rashes.
Clinical Impact and Efficacy Data
The integration of AKT inhibitors represents a massive paradigm shift in managing treatment-refractory disease, particularly in advanced breast cancer. Historically, advanced HR+/HER2- breast cancer is managed with endocrine (hormone) therapy. Over time, the tumor mutates to survive without hormones. Clinical data confirms that the hyperactive PI3K/AKT/mTOR pathway is the primary escape route the cancer utilizes to bypass hormone therapy.
Landmark Trial Metrics
The regulatory approval of targeted drugs relies on massive, randomized Phase III clinical trials, monitored by organizations like the American Society of Clinical Oncology (ASCO). When evaluating an AKT inhibitor (such as capivasertib in the landmark CAPItello-291 trial), oncologists evaluate specific survival metrics:
- Progression-Free Survival (PFS): This metric measures the duration a patient lives with the disease without radiographic evidence of tumor progression. In patients harboring confirmed PIK3CA, AKT1, or PTEN alterations, the addition of an AKT inhibitor to standard endocrine therapy (fulvestrant) demonstrated a statistically significant extension in median PFS. Clinical data illustrates that the targeted combination effectively doubled the time patients lived without their disease progressing compared to endocrine therapy alone.
- Objective Response Rate (ORR): This represents the percentage of patients whose tumors shrink by a predetermined, medically significant volume. Combination therapy yielded a substantially higher ORR, confirming the inhibitor’s ability to force measurable tumor regression in heavily pre-treated, endocrine-resistant cohorts.
Visualizing Clinical Data: Understanding Trial Metrics
Medical literature utilizes specific data visualizations to illustrate clinical trial results. Patients reviewing primary source research regarding their specific treatment protocols will frequently encounter these standardized graphs.
The Kaplan-Meier Survival Curve
A Kaplan-Meier curve tracks the percentage of patients whose disease remains strictly controlled over a defined period.
- The Axes: The horizontal x-axis measures time (usually in months), starting from the initiation of therapy. The vertical y-axis represents the percentage of patients (from 100% down to 0%) who have not experienced a progression event.
- The Clinical Gap: The graph features descending “stair-step” lines comparing the control group (standard therapy) against the experimental group (AKT inhibitor therapy). In a successful targeted therapy trial, the line representing the inhibitor remains significantly higher and stretches further to the right. The wide visual gap between the two curves represents the exact clinical benefit: the extended months of strict disease control provided by the drug.

The Genomic Heatmap
A heatmap is a visual matrix used to display patterns of DNA mutations across thousands of sequenced patients.
- The Layout: Each column represents a single patient, while each row represents a specific gene (e.g., AKT1, PIK3CA, PTEN). Colored blocks indicate the presence of a pathological mutation.
- The Data: In an AKT-focused heatmap, the data visually confirms that mutations in the PI3K/AKT/PTEN pathway are typically mutually exclusive. If a patient harbors an AKT1 mutation, they rarely possess a concurrent PIK3CA mutation. This demonstrates to researchers that a tumor only needs to break a single link in this specific signaling chain to achieve runaway, malignant growth.

Precision Medicine & Acquired Resistance
Treating a malignancy based on an AKT1 E17K mutation or a PTEN deletion is the strict definition of Precision Medicine. These alterations cannot be identified via standard CT imaging or basic histological stains; they require comprehensive molecular profiling. An oncologist must order Next-Generation Sequencing (NGS) of the tumor tissue or a liquid biopsy to sequence the tumor’s DNA and isolate the exact genomic error.
Addressing Drug Resistance
Precision oncology requires anticipating biological adaptation. While AKT inhibitors are highly effective, the tumor will eventually attempt to bypass the therapeutic blockade, leading to acquired resistance. This resistance manifests through well-documented biological mechanisms:
- Loss of Feedback Inhibition: Silencing AKT removes a natural cellular feedback loop. In response to the drug, the starving tumor cell aggressively upregulates upstream surface receptors (such as HER3, IGF-1R, or EGFR) to try and force the blocked pathway back open.
- Compensatory Pathway Activation: The cellular network is highly interconnected. If the PI3K/AKT highway is permanently stalled, the tumor alters its genetic expression to rely on parallel signaling networks, such as the RAS/MEK/ERK cascade. The cancer essentially builds a new cellular road around the roadblock to resume proliferation.
When clinical resistance occurs, oncologists typically conduct a subsequent biopsy to map the tumor’s newly adapted genomic profile. Understanding exactly how the cancer bypassed the drug dictates the next line of treatment. Currently, extensive clinical trials are actively evaluating combination regimens—such as pairing an AKT inhibitor with a MEK inhibitor—to enact a dual-blockade and prevent these escape routes before they can initiate.psy to map the tumor’s adapted genomic profile. Current clinical trials are actively evaluating combination regimens—such as pairing an AKT inhibitor with a MEK inhibitor or specific receptor degraders—to enact a dual-blockade and prevent these escape routes.
People Also Ask
What does it mean to have an AKT mutation in cancer?
An AKT mutation is a specific genetic error (often the AKT1 E17K alteration) within a tumor’s DNA. It causes the AKT protein—a central regulator of cell survival—to become permanently stuck in an active state. This constant activation instructs the cancer cells to multiply rapidly, consume excess energy, and evade natural cell death (apoptosis), directly driving tumor progression and causing resistance to standard hormone therapies.
Are AKT mutations hereditary and passed down to children?
No. The AKT1 mutations driving solid tumors are strictly “somatic” mutations. This means the genetic alteration is acquired during your lifetime specifically within the localized tumor cells due to DNA copying errors or environmental factors. It is not a “germline” mutation, meaning it is not present in your reproductive cells and cannot be passed down to your children.
How does my oncologist test for an AKT pathway alteration?
AKT mutations cannot be detected using standard CT scans or basic histological tissue stains; they require comprehensive molecular profiling. Your oncology team will order Next-Generation Sequencing (NGS) on a physical tissue biopsy or a liquid biopsy (a specialized blood test that detects circulating tumor DNA). This genomic sequencing maps the tumor’s exact molecular signature to confirm the alteration.
Which cancers most commonly harbor AKT mutations?
While AKT pathway hyperactivation occurs across many malignancies, direct AKT1 point mutations are most prominently identified in Hormone Receptor-positive (HR+), HER2-negative advanced breast cancers, appearing in roughly 4% to 8% of cases. Broader pathway alterations—including PTEN deletions or PIK3CA mutations—are also highly prevalent in gynecologic cancers (endometrial and ovarian) and metastatic castration-resistant prostate cancer (mCRPC).
How do AKT inhibitors work compared to traditional chemotherapy?
Traditional chemotherapy is cytotoxic, indiscriminately attacking all rapidly dividing cells in the body, leading to severe systemic side effects. AKT inhibitors (like capivasertib) are highly targeted, small-molecule therapies. They are engineered to physically bind to the defective AKT protein, blocking its cellular fuel source (ATP). By silencing this specific hyperactive engine, the drug arrests tumor division while largely sparing healthy tissue.
Why do targeted AKT inhibitors cause high blood sugar (hyperglycemia)?
This is an expected, on-target effect. The AKT2 protein isoform plays a critical role in normal human insulin signaling. When you eat, insulin activates AKT2 to pull glucose from your blood into your cells. Because pan-AKT inhibitors block all AKT isoforms globally, they temporarily induce peripheral insulin resistance. Oncologists proactively manage this expected hyperglycemia with routine monitoring, dietary modifications, and standard anti-diabetic medications like metformin.
What is the clinical relationship between AKT, PIK3CA, and PTEN?
AKT, PIK3CA, and PTEN are all critical components of the same cellular signaling highway (the PI3K/AKT/mTOR pathway). PIK3CA acts as the upstream trigger, AKT is the central communication hub, and PTEN acts as the physiological brake. A mutation in any of these three genes achieves the exact same pathological result: pathway hyperactivation. Notably, a tumor usually only needs to break one of these links to drive malignant growth.
Can the tumor develop resistance to an AKT inhibitor over time?
Yes, cancer is biologically highly adaptive. Over time, the tumor may bypass the therapeutic blockade by activating parallel signaling networks (such as the RAS/MEK/ERK pathway) or upregulating upstream receptors to resume proliferation. When clinical resistance occurs, oncologists will perform a subsequent biopsy to map the adapted genomic profile and determine the next line of treatment, which may include combination targeted therapies.
Are clinical trials a viable option for AKT-mutated cancers?
Yes. In precision oncology, clinical trials are not viewed as a “last resort.” They frequently offer front-line access to next-generation therapies. Researchers are currently evaluating powerful combination regimens—such as pairing AKT inhibitors with targeted protein degraders or MEK inhibitors—to enact dual-blockades that prevent the tumor from developing acquired resistance. If your oncologist suggests a trial, it means they believe it may provide a superior clinical outcome.
Disclaimer: The information provided in this article is for educational and informational purposes only. It is not a substitute for professional medical advice, diagnosis, or treatment. Always consult with your oncologist, hematologist, or healthcare provider regarding your specific medical condition, genetic testing results, and customized treatment plan. Medically Reviewed by DR. Salma Elreedy, MD (Clinical Oncology).
Educational References
The biological mechanisms, genetic data, and clinical trial statistics detailed in this comprehensive guide are grounded in rigorous, peer-reviewed science published by authoritative medical institutions. For primary source data and detailed clinical guidelines, consult the following resources:
The National Center for Biotechnology Information (NCBI) / PubMed
The American Society of Clinical Oncology (ASCO)