Sourcing Disclosure: Licensed Bangladesh Global Sourcing Specialist facilitating global access under Named Patient Regulations.
Expert-reviewed clinical data for prescription-only medicine.
Clinical Guide to the FLT3 Mutation in Acute Myeloid Leukemia (AML)
Table of Content
A primary diagnosis of Acute Myeloid Leukemia (AML) introduces patients and their families to a highly complex landscape of hematology, molecular biology, and genomic profiling. Modern precision oncology no longer relies solely on identifying the morphological type of cancer present in the bone marrow. Instead, the standard of care requires identifying the specific genetic anomalies and molecular drivers powering the leukemic cells.
One of the most frequently identified and clinically consequential genetic biomarkers in Acute Myeloid Leukemia is the FLT3 mutation. If your bone marrow biopsy or peripheral blood pathology report indicates the presence of an FLT3 mutation, your hematology team has pinpointed the primary biological mechanism driving your disease. This specific genomic finding is highly actionable and directly dictates your subsequent treatment protocols. This comprehensive guide details the cellular biology of the FLT3 gene, the exact mechanisms by which targeted pharmaceutical therapies disrupt the disease, and what this genetic marker ultimately means for your clinical prognosis.
The Biology of the FLT3 Receptor
To comprehend the mutation, it is necessary to understand normal blood cell production. Hematopoiesis—the continuous manufacturing of blood cells—occurs within the bone marrow cavity. This biological manufacturing process must remain under strict regulatory control. The human body requires a precise equilibrium: adequate white blood cells for immunological defense, sufficient red blood cells for oxygen transport, and adequate platelets for hemostasis (blood clotting).
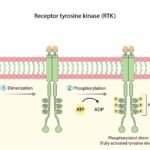
The FLT3 gene (FMS-like tyrosine kinase 3) provides the physiological blueprint for constructing the FLT3 protein. This protein functions as a critical receptor kinase located on the external cellular membrane of early, multipotent hematopoietic stem cells (immature blood cells).
In a physiologically healthy bone marrow microenvironment, the FLT3 receptor serves as a primary regulatory switch for cellular proliferation and differentiation.
- The Inactive State: When physiological blood counts are stable and adequate, the receptor remains entirely unactivated. The hematopoietic stem cell remains in a quiescent, resting phase.
- The Active State: When the physiological system demands the generation of new blood cells, an extracellular chemical messenger known as the FLT3 ligand binds directly to the receptor. This binding event triggers a brief, highly controlled intracellular signal, commanding the immature stem cell to undergo maturation, proliferate, and eventually mobilize into the peripheral bloodstream. Following this sequence, the activation signal is biologically terminated.
The Pathogenesis of the FLT3 Alteration
When a pathological alteration occurs within the sequence of the FLT3 gene, the regulatory mechanisms fail. The mutated DNA instructions force the cell to synthesize a structurally defective, continuously active FLT3 receptor.
Instead of remaining quiescent until bound by the extracellular ligand, this structurally deformed receptor undergoes constitutive activation. It is permanently locked in an activated conformation. The stem cell receives an unrelenting, unregulated command to divide and proliferate. Furthermore, because this signaling is biochemically abnormal, these cells fail to undergo standard differentiation. They never mature into functional, mature blood components. They arrest in their development, remaining as immature, non-functional cells termed leukemic blasts. As these malignant blasts hyper-proliferate, they physically crowd out healthy hematopoietic cells within the marrow space, precipitating the severe, systemic symptoms characteristic of acute leukemia.
Genomic Classifications: FLT3-ITD and FLT3-TKD
FLT3 mutations represent the most prevalent genetic abnormality identified in Acute Myeloid Leukemia, presenting in approximately 30% of all adult AML diagnoses. Clinical diagnostics broadly categorize these mutations into two distinct structural variants.
1. FLT3-ITD (Internal Tandem Duplication) This variant is the most common and clinically aggressive presentation, accounting for roughly 25% of all adult AML cases.
- The Genetic Mechanism: During DNA replication, a specific sequence of the genetic code is erroneously duplicated and inserted sequentially next to the original DNA sequence.
- The Biological Consequence: This tandem duplication predominantly manifests within the juxtamembrane domain of the receptor. In normal physiology, this domain functions as a structural autoinhibitory mechanism—essentially a biological safety latch that prevents unprompted activation. The aberrant duplication physically elongates and disrupts this domain, destroying the autoinhibitory latch. Consequently, the receptor kinase continuously fires proliferation signals independent of natural regulatory controls.
2. FLT3-TKD (Tyrosine Kinase Domain Point Mutation) This secondary variant is statistically less common, presenting in approximately 5% to 7% of AML cases.
- The Genetic Mechanism: Rather than a sequence duplication, the TKD variant involves a single nucleotide polymorphism (a missense mutation). A single biochemical letter in the DNA sequence is erroneously substituted. The most frequently observed point mutation occurs at the D835 hotspot.
- The Biological Consequence: This point mutation occurs directly within the active kinase domain—the catalytic engine room of the receptor. The specific amino acid substitution alters the three-dimensional conformation of the activation loop, permanently stabilizing the receptor in an open, active state, driving continuous oncogenic signaling.
Intracellular Signaling: The Biochemical Cascade
To arrest leukemic proliferation, oncologists must map the precise intracellular communication networks exploited by the mutated receptor. This network is referred to as the signal transduction pathway.
Upon constitutive activation, the mutated FLT3 receptor initiates an aggressive internal biochemical cascade, transferring energy via autophosphorylation down several distinct cellular pathways:
- The STAT5 Pathway: The FLT3 receptor directly phosphorylates the STAT5 protein, which translocates to the cellular nucleus. Here, it upregulates anti-apoptotic genes, overriding the cell’s natural life-cycle limitations and aggressively preventing programmed cell death (apoptosis).
- The RAS/MAPK Pathway: The receptor activation engages a subsequent cascade of proteins, including RAS, RAF, MEK, and ERK. This specific kinase chain delivers the terminal command for unchecked cellular division and mitosis.
- The PI3K/AKT Pathway: This secondary signaling route optimizes cellular metabolism and reinforces survival directives, ensuring the leukemic blast population thrives even under adverse physiological conditions.
Mechanism of Action: Tyrosine Kinase Inhibitors (TKIs)
Historically, the sole therapeutic approach for AML relied on intensive, non-selective systemic chemotherapy regimens. While cytotoxic chemotherapy remains a critical pillar of induction therapy, pharmaceutical science has revolutionized the standard of care through the development of targeted FLT3 Inhibitors (such as midostaurin, gilteritinib, and quizartinib).
Classified as Tyrosine Kinase Inhibitors (TKIs), these small-molecule therapeutics are engineered with atomic precision to inhibit the defective enzymatic engine.
- Target Binding: Following systemic administration, the medication circulates and permeates the leukemic blasts within the bone marrow. The molecular structure of the inhibitor is designed to interface directly with the intracellular kinase domain of the mutated FLT3 protein.
- Competitive Inhibition: The mutated FLT3 engine relies on adenosine triphosphate (ATP) as the fundamental cellular energy source to fuel its phosphorylation cascades. The TKI molecule is structurally synthesized to perfectly occupy the ATP-binding cleft within the receptor. By acting as a physical wedge, the drug competitively blocks ATP from accessing the kinase domain.
- Signal Termination: Deprived of its biochemical fuel, the enzymatic activity of the FLT3 receptor is immediately neutralized. The receptor ceases transmission down the STAT5, RAS/MAPK, and PI3K/AKT pathways. Abruptly severed from these mandatory survival and proliferation signals, the leukemic blasts undergo rapid, systemic apoptosis.
Target Specificity and Systemic Impact
The fundamental objective of targeted therapy is maximizing target specificity—the pharmacological ability to eradicate malignant cells while minimizing cytotoxicity to healthy tissues.
FLT3 inhibitors demonstrate high specificity because normal solid organ tissues (hepatic, cardiac, pulmonary) do not exhibit functional dependence on the FLT3 pathway for cellular survival. The medication largely bypasses these organ systems without engagement. However, normal, healthy hematopoietic stem cells residing in the marrow also express standard FLT3 receptors. Because the inhibitors block FLT3 signaling universally, therapeutic administration will temporarily suppress normal blood cell production. This anticipated clinical effect, known as myelosuppression, manifests as profound neutropenia, anemia, and thrombocytopenia. Hematologists proactively manage this suppression through rigorous transfusion support and prophylactic antimicrobial therapy while the targeted agents clear the leukemic burden.
Clinical Trial Outcomes and Prognostic Impact
The integration of FLT3-targeted therapies represents a paradigm shift in hematological oncology. Historically, the presence of an FLT3-ITD mutation conferred a highly unfavorable prognosis, characterized by aggressive relapse rates following standard cytarabine-based chemotherapy. The deployment of precision TKIs has fundamentally improved survival metrics.
First-Line Treatment Integration (The RATIFY Trial)
For newly diagnosed patients harboring an FLT3 mutation, current clinical protocols frequently combine the first-generation inhibitor midostaurin with standard induction and consolidation chemotherapy. The landmark RATIFY trial definitively established that integrating the FLT3 inhibitor alongside standard cytotoxic regimens significantly improved overall survival compared to cohorts receiving chemotherapy alone, lowering the aggregate risk of mortality by 22%.
 1")
Relapsed or Refractory Interventions (The ADMIRAL Trial) In clinical scenarios where the leukemia returns (relapse) or demonstrates primary resistance to initial induction (refractory), highly potent, second-generation inhibitors such as gilteritinib are deployed. Data from the ADMIRAL trial demonstrated that gilteritinib, utilized as a single-agent therapy, achieved substantially higher complete remission rates and doubled the median overall survival time compared to standard salvage chemotherapy in this highly challenging patient population.
 2")
Bridging to Allogeneic Stem Cell Transplant For many clinically eligible patients with FLT3-mutated AML, the ultimate therapeutic objective is achieving a sufficiently deep molecular remission to permit an allogeneic hematopoietic stem cell transplant. FLT3 inhibitors demonstrate high efficacy in suppressing the leukemic blast volume (minimal residual disease) to the threshold required to make this potentially curative cellular therapy feasible.
Understanding Clinical Survival Plots
In hematological literature, clinical efficacy is visually quantified to demonstrate the statistical benefit of targeted interventions. The most frequently utilized metric is the Kaplan-Meier survival curve.
- The Axes: The horizontal x-axis represents temporal progression (measured in months or years from trial initiation). The vertical y-axis measures the cumulative percentage of surviving patients within the cohort.
- Curve Interpretation: The graph plots descending step-functions for distinct patient groups (e.g., standard chemotherapy vs. targeted FLT3 inhibition). In trials demonstrating high efficacy, the curve representing the targeted therapy cohort remains visibly elevated and extends further along the temporal axis before descending. The distinct visual separation between the curves illustrates the direct, statistically significant extension of life provided by the targeted therapeutic agent.
 3")
The Challenge of Acquired Drug Resistance
While FLT3 TKIs are highly effective at inducing morphological and molecular remissions, leukemic biology is characterized by aggressive clonal evolution. Over time, malignant cells frequently undergo further genomic alterations to bypass the pharmaceutical blockade, a phenomenon known as acquired drug resistance. When blast counts inevitably begin to rise despite targeted therapy, the hematology team must evaluate for secondary resistance mechanisms.
On-Target Resistance (Gatekeeper Mutations) The leukemic clones may acquire secondary point mutations directly within the FLT3 kinase domain. The most clinically significant is the F691L “gatekeeper” mutation. This specific amino acid substitution physically alters the three-dimensional topography of the drug-binding pocket, creating steric hindrance that actively repels inhibitors like midostaurin and quizartinib, rendering the leukemic cells completely refractory to the drug.
Off-Target Resistance (Bypass Signaling) Alternatively, the malignant cells may entirely circumvent the FLT3 pathway. The leukemic clones activate parallel genomic instructions, upregulating alternative survival networks such as the AXL kinase pathway or acquiring downstream RAS and MAPK mutations. This adaptive bypass completely circumvents the blocked FLT3 receptor, reigniting leukemogenesis.
To combat resistance, clinical protocols increasingly focus on next-generation inhibitors designed to overcome gatekeeper mutations, as well as rational combination therapies (such as combining FLT3 inhibitors with BCL-2 inhibitors like venetoclax) to simultaneously blockade multiple cellular pathways and prevent the emergence of resistant clones.
Educational References
The biological mechanisms, genetic data, and clinical trial statistics detailed in this comprehensive guide are grounded in rigorous, peer-reviewed science published by authoritative medical institutions. For primary source data and detailed clinical guidelines, consult the following resources:
The National Center for Biotechnology Information (NCBI) / PubMed
The American Society of Clinical Oncology (ASCO)
The National Cancer Institute (NCI)
The Cancer Genome Atlas (TCGA)
Disclaimer: The information provided in this article is for educational and informational purposes only. It is not a substitute for professional medical advice, diagnosis, or treatment. Always consult with your oncologist, hematologist, or healthcare provider regarding your specific medical condition, bone marrow biopsy results, and customized treatment plan. Medically Reviewed by DR. Salma Elreedy, MD (clinical Oncology), Sphinx Cure Oncology Center.