Prescription required. We facilitate access to medicines already prescribed by your treating physician. We do not provide medical advice or diagnoses.
KRAS G12C Mutation in Advanced NSCLC: Targeted Therapies and Clinical Management
A cancer diagnosis often introduces a completely new vocabulary. You might hear your oncology team discuss genes, mutations, and targeted therapies. In modern oncology, treating cancer requires understanding the specific genetic blueprint of the tumor. One of the most significant discoveries in cancer genetics is the KRAS G12C mutation.
If your biopsy or blood test report indicates a KRAS G12C mutation, your medical team has identified the specific biological mechanism driving your cancer. This knowledge directly influences your treatment options. This guide explains the science behind this mutation, how new medications target it, and what it means for your prognosis.
The Basics: What is the KRAS Gene?
To understand the mutation, we first need to look at normal cell biology. The human body is made of trillions of cells. Inside almost every cell is DNA, which acts as an instruction manual. Genes are specific sections of DNA that tell the cell how to make proteins.
The KRAS gene provides instructions for making the KRAS protein. In a healthy cell, the KRAS protein acts like a light switch that controls cell growth and division.
- The “ON” Position: When the body needs new cells to replace old or damaged ones, signals from outside the cell flip the KRAS switch “ON.” The cell begins to divide.
- The “OFF” Position: Once enough cells are produced, the KRAS protein automatically flips itself back to the “OFF” position. The cell stops dividing.
This precise on-and-off mechanism keeps tissue growth balanced and healthy.
The Broken Light Switch Analogy
When a mutation occurs in the KRAS gene, the instruction manual has a misprint. The cell builds a defective KRAS protein. In the case of cancer, this defective protein loses its ability to turn itself off.
The switch gets permanently stuck in the “ON” position. The cell receives a constant, uninterrupted command to divide, multiply, and survive. These cells rapidly accumulate, eventually forming a tumor. For decades, researchers knew this broken switch caused cancer, but they lacked the technology to fix it or block it.
The Science of the Mutation: At the DNA Level
To understand exactly what goes wrong, we must look at the genetic code. DNA is written using four chemical bases: Adenine (A), Cytosine (C), Guanine (G), and Thymine (T). The cell reads these letters in groups of three, called a codon. Each codon tells the cell to use a specific building block, called an amino acid, to build a protein.
In a KRAS G12C mutation, a specific error happens at Codon 12 of the KRAS gene.
- The Normal State: In a healthy KRAS gene, the DNA sequence at Codon 12 tells the cell to use an amino acid called Glycine (represented by the letter “G”). Glycine is small and flexible, allowing the KRAS protein to function normally and turn itself off.
- The Mutation State: The DNA experiences a single spelling error. A “G” is swapped for a “T” or “A” in the genetic code. This misprint tells the cell to insert a completely different amino acid called Cysteine (represented by the letter “C”) instead of Glycine.
This specific change—from Glycine at position 12 to Cysteine—is why it is named KRAS G12C.
Cysteine is larger and more chemically reactive than Glycine. This single substitution alters the physical shape of the KRAS protein, destroying its internal shut-off mechanism.
Identifying the Mutation Hotspots
Mutations do not happen evenly across all cancers. The KRAS G12C mutation typically occurs in specific types of tumors, often referred to as mutation “hotspots.” According to data from The Cancer Genome Atlas (TCGA) and the National Cancer Institute (NCI):
- Non-Small Cell Lung Cancer (NSCLC): This is the most common location for the KRAS G12C mutation. Overall, it is found in approximately 13% to 15% of all lung adenocarcinomas and is strongly associated with a history of tobacco smoking. However, real-world clinical data shows a massive geographic and ethnic variance in its prevalence:
- North America & Europe: Highly prevalent, appearing in roughly 11% to 16% of NSCLC patients.
- Latin America: Present in approximately 7% to 9% of patients.
- Asia: Significantly lower, appearing in only 1% to 4% of NSCLC patients (in these populations, EGFR mutations are the dominant genetic driver).
- Colorectal Cancer (CRC): The mutation is found in about 3% to 4% of colorectal cancers.
- Other Solid Tumors: It appears in less than 1% to 2% of pancreatic cancers, appendiceal cancers, and other rare tumor types.
Pathway & Connectivity: The Biological Chain Reaction
To understand how the mutation builds a tumor, and how drugs stop it, we need to trace the Signaling Pathway. Cells communicate through internal chain reactions, similar to a relay race.
The specific route KRAS uses is called the RAS/MAPK signaling cascade.
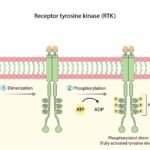
- The Receptors: On the outside surface of the cell, there are receiver antennas (such as the EGFR receptor). They wait for growth signals from the body.
- The Relay Hand-off (KRAS): When a signal arrives, the receptor activates KRAS just inside the cell membrane.
- The Chain Reaction: KRAS (the manager) passes the signal to a protein called RAF. RAF passes it to MEK. MEK passes it to ERK.
- The Final Command: ERK travels directly into the nucleus of the cell (the control center) and delivers the final command: “Start dividing and do not die.”
When KRAS G12C is present, the relay race never waits for a starting signal from the receptors. The mutated KRAS protein continuously fires the signal down to RAF, MEK, and ERK. The cell divides endlessly. To stop the tumor, medical treatments must break this chain reaction.
Mechanism of Action: How the Inhibitor Works
For over forty years, KRAS was labeled “undruggable.” The protein has a smooth, spherical shape, lacking the deep pockets or grooves that medications normally use to latch onto a target.
However, scientists discovered a hidden vulnerability specific to the G12C mutation. The abnormal Cysteine amino acid creates a tiny, shallow pocket on the surface of the protein (known as the Switch-II pocket) that does not exist in healthy KRAS proteins.
This discovery led to the development of KRAS G12C inhibitors, such as sotorasib and adagrasib. These are small-molecule, oral medications engineered to exploit this exact structural flaw.
Finding and “Turning Off” the Cancer Cells
- Target Recognition: After the patient swallows the pill, the drug enters the bloodstream and travels to the tumor. The chemical structure of the drug is designed to search specifically for the mutant Cysteine residue.
- Covalent Binding: Once the drug finds the mutated Cysteine, it forms a strong chemical bond (a covalent bond) with it. It acts like a key snapping permanently into a lock.
- Locking the Switch: By attaching to this specific pocket, the drug physically forces the entire KRAS protein to change its shape. It traps the protein in its inactive, “OFF” state.
- Halting the Chain Reaction: With KRAS permanently locked in the “OFF” position, it can no longer send signals to RAF, MEK, or ERK. The relay race stops. The cancer cell stops receiving the command to grow and eventually undergoes programmed cell death (apoptosis).
Target Specificity: Sparing Healthy Cells
Traditional chemotherapy attacks any rapidly dividing cell in the body, which leads to widespread side effects like hair loss, nausea, and low white blood cell counts.
KRAS G12C inhibitors work differently because of target specificity. Healthy cells rely on the normal KRAS protein, which contains Glycine, not Cysteine. Because the inhibitor drug is chemically programmed to only bond with Cysteine, it ignores normal KRAS proteins. The drug passes by healthy tissue without attaching.
While targeted therapies still have side effects (such as gastrointestinal issues or elevated liver enzymes as the body processes the medication), they generally avoid the severe systemic toxicity of traditional chemotherapy because they selectively target the genetic error unique to the tumor.
Clinical Impact & Prognosis
The introduction of KRAS G12C inhibitors has fundamentally changed the treatment landscape for patients harboring this specific mutation.
Previously, patients with KRAS-mutated lung cancer had limited options once the disease progressed past initial chemotherapy and immunotherapy. The mutation was considered a marker for a more aggressive disease course. Today, identifying this mutation opens the door to personalized, targeted treatment that can significantly alter a patient’s prognosis.
Summarizing Clinical Trial Data
The approval and use of these drugs are based on rigorous clinical trials monitored by organizations like the American Society of Clinical Oncology (ASCO). The data evaluates how well the drugs shrink tumors and extend life.
When evaluating these drugs, oncologists look at specific metrics:
- Objective Response Rate (ORR): This represents the percentage of patients whose tumors shrink by a predetermined, medically significant amount. In major trials for KRAS G12C inhibitors (like the CodeBreaK and KRYSTAL trials) used in previously treated non-small cell lung cancer, the ORR generally ranges from 36% to 43%. This means nearly 4 out of 10 patients experience substantial tumor shrinkage.
- Disease Control Rate (DCR): This includes patients whose tumors shrank, plus patients whose tumors stopped growing and remained stable. The disease control rate for these inhibitors frequently exceeds 80%. The medication successfully halts the progression of the cancer in the vast majority of patients.
- Progression-Free Survival (PFS): This measures the length of time during and after treatment that a patient lives with the disease without it worsening. Clinical data shows a median progression-free survival of approximately 6.5 to 6.8 months, with some patients experiencing durable responses lasting well beyond a year.
- Overall Survival (OS): This measures the total length of survival from the start of treatment. Current data shows median overall survival rates extending past 12 months for patients who had already exhausted other standard treatment options.
These statistics represent a substantial clinical improvement over historical outcomes for advanced, pre-treated lung cancer, offering patients more time with controlled disease.
Visualizing the Data: Survival Plots and Heatmaps
Medical literature often uses specific graphs to illustrate clinical trial results. If you read research papers regarding KRAS G12C, you will likely encounter two types of data visualization.
The Kaplan-Meier Survival Plot
A Kaplan-Meier curve tracks the percentage of patients surviving or living without disease progression over time.

- The Vertical Axis (Y-axis): This line up the side represents the percentage of patients (from 0% to 100%).
- The Line (The Curve): The graph starts at 100% on the top left. As time passes and patients experience disease progression, the line steps downward.
When you look at a survival plot for a KRAS G12C inhibitor compared to standard chemotherapy, you will see two lines. The line representing the targeted inhibitor will stay higher and stretch further to the right before stepping down. This visual “gap” between the two lines represents the clinical benefit: a higher percentage of patients remaining stable for a longer duration.
The Waterfall Plot
A Waterfall Plot is used to show tumor shrinkage.
- The middle horizontal line represents a 0% change in tumor size (the baseline).
- Each vertical bar represents a single patient in the clinical trial.
- Bars reaching above the zero line indicate tumors that grew.
- Bars extending below the zero line indicate tumors that shrank.
In a successful trial for a KRAS G12C inhibitor, the graph looks like a waterfall flowing downward. The vast majority of the vertical bars point downwards, visually demonstrating that most patients experienced some level of tumor reduction.
Precision Medicine & Resistance
The treatment of KRAS G12C mutations is a textbook example of Precision Medicine (also called personalized medicine).
Precision medicine moves away from treating cancer based solely on its location in the body (e.g., “lung cancer”). Instead, it treats the cancer based on its specific genetic driver. A patient will not receive a KRAS G12C inhibitor unless a molecular biopsy (Next-Generation Sequencing of tumor tissue or a liquid blood biopsy) confirms the exact G12C spelling error exists. The treatment is personalized to the molecular signature of the tumor.
Addressing Drug Resistance
While targeted therapies are highly effective, cancer is biologically adaptable. Over time, the tumor may find a way to survive despite the medication. This is known as Drug Resistance.
When a KRAS G12C inhibitor eventually stops working, it is usually because the cancer cells have evolved. Resistance generally occurs through two main mechanisms:
- On-Target Resistance (Changing the Lock): The cancer cell develops a secondary mutation directly on the KRAS protein. For example, a new mutation called Y96D might appear. This changes the physical shape of the Switch-II pocket. The drug’s “key” no longer fits into the lock, and the drug physically cannot bind to the KRAS protein anymore.
- Off-Target Resistance (Taking a Detour): The tumor bypasses the KRAS blockade entirely. The cancer cell amplifies other signaling pathways. It might increase the number of EGFR receptors on the cell surface, or it might activate alternative proteins like BRAF or MET. The cancer builds a new cellular road around the roadblock, restarting the relay race and the growth signals.
When resistance develops, your medical team will likely order a new biopsy to map the tumor’s new genetic profile. Understanding exactly how the cancer adapted dictates the next line of treatment.
Currently, extensive clinical trials are underway evaluating combination therapies. Researchers are testing KRAS G12C inhibitors combined with other targeted drugs (like EGFR inhibitors or MEK inhibitors) to block the primary pathway and the detour pathways simultaneously, aiming to delay or prevent resistance from occurring.
Authority & Trust: Educational References
The scientific explanations, statistical ranges, and biological mechanisms detailed in this guide are synthesized from peer-reviewed literature and data published by authoritative oncological and genomic organizations.
For further reading and primary source data, patients and families can consult:
- The National Cancer Institute (NCI): Provides comprehensive overviews of targeted cancer therapies and the RAS/MAPK signaling pathway.
- The American Society of Clinical Oncology (ASCO): Publishes current clinical practice guidelines and primary clinical trial results regarding the efficacy of KRAS inhibitors.
- The National Center for Biotechnology Information (NCBI): Hosts PubMed, a repository of peer-reviewed medical journals detailing the molecular mechanisms of KRAS G12C binding and acquired drug resistance.
- The Cancer Genome Atlas (TCGA): A landmark cancer genomics program that cataloged the frequency and genetic characteristics of mutations like KRAS G12C across multiple cancer types.
Disclaimer: The clinical information, genomic data, and treatment protocols detailed on genericoncology.com regarding the KRAS G12C mutation and advanced NSCLC are provided strictly for educational and informational purposes. This content does not constitute professional medical advice, a formal diagnosis, or a specific treatment recommendation, nor does reading this guide establish a doctor-patient relationship. Medically Reviewed by (Dr. Salma Elreedy), clinical oncology, Sphinx Cure Oncology Center