Sourcing Disclosure: Licensed Bangladesh Global Sourcing Specialist facilitating global access under Named Patient Regulations.
Expert-reviewed clinical data for prescription-only medicine.
CDK4/6 Pathway Dysregulation in Oncology: Molecular Drivers and Targeted Inhibitor Protocols
Protocol Summary
Receiving an advanced oncology diagnosis necessitates an immediate transition from generalized anatomical staging to precise molecular and genomic profiling. In contemporary clinical oncology, the standard of care dictates that malignancies are evaluated and treated based on their specific, microscopic biological drivers. One of the most critical regulatory mechanisms identified in human cellular biology involves the Cyclin-Dependent Kinases 4 and 6 (CDK4/6) pathway.
When comprehensive genomic profiling (CGP), immunohistochemistry (IHC), or next-generation sequencing (NGS) identifies a CDK4/6 pathway alteration—or when clinical guidelines dictate the deployment of a CDK4/6 inhibitor—oncologists are directly targeting the core catalytic engine of cellular division. Comprehending the molecular function of these specific kinases is essential for understanding the rationale behind personalized treatment sequencing. This monograph details the molecular biology of the CDK4/6 cell cycle pathway, the pharmacological mechanism of selective kinase inhibitors, and the clinical implications for patients navigating advanced malignancies, particularly Hormone Receptor-positive (HR+) breast cancer.
The Molecular Biology of the Cell Cycle and the G1/S Checkpoint
To accurately conceptualize tumor pathology, one must first examine the regulatory mechanisms governing the normal cellular life cycle. Somatic cells undergo a highly conserved, strictly regulated sequence of events culminating in cellular division, known as the cell cycle. This cycle is delineated into distinct phases: G1 (Gap 1), S (Synthesis), G2 (Gap 2), and M (Mitosis).
Progression through these phases is mediated by highly stringent internal security checkpoints. The most critical regulatory juncture is the G1/S transition, known as the restriction point. Passage through this checkpoint commits the cell to DNA replication and subsequent division.
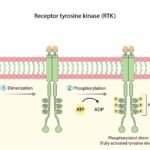
The CDK4 and CDK6 genes encode highly specific serine/threonine kinases. Under baseline physiological conditions, these kinases remain catalytically inactive. Activation is strictly dependent upon the presence of mitogenic signals (external growth factors) which stimulate the transcription and synthesis of D-type cyclins (Cyclin D1, D2, and D3).
When Cyclin D binds to CDK4 or CDK6, it forms an active holoenzyme complex. This Cyclin D-CDK4/6 complex is the primary catalytic driver required to push the cell past the restriction point. Once the cell successfully replicates its DNA and divides, the complex is degraded, and the cell returns to a quiescent (G0) state or prepares for another highly regulated cycle.
Genomic Alterations Driving CDK4/6 Hyperactivation
Malignancies do not merely exhibit a loss of structure; they represent a fundamental hijacking of the cell cycle machinery. In CDK4/6-driven tumors, genomic instability leads to the permanent, constitutive activation of this pathway. The tumor cell becomes untethered from external mitogenic requirements, receiving a continuous, uninterrupted biological command to proliferate.
This hyperactivation is rarely driven by isolated point mutations within the CDK4/6 genes themselves. Instead, it manifests through severe structural imbalances, locus amplifications, or the deletion of critical tumor suppressor genes within the immediate molecular environment.
1. Gene Amplification and Overexpression
In many solid tumors, genomic instability results in the focal amplification of specific chromosomal regions. The cell produces massively disproportionate quantities of specific pathway proteins.
- CCND1 (Cyclin D1) Amplification: Highly prevalent in HR+ breast cancers and squamous cell carcinomas, the overproduction of Cyclin D1 forces continuous binding with available CDK4/6 proteins, maintaining the kinases in a perpetually active state.
- CDK4 Amplification: Malignancies such as well-differentiated liposarcomas and certain glioblastomas frequently exhibit the direct genomic amplification of the CDK4 locus, saturating the intracellular environment with hyperactive catalytic domains.
2. Loss of Tumor Suppressor CDKN2A (p16INK4A)
Healthy cellular physiology relies on endogenous inhibitory proteins to prevent uncontrolled proliferation. The CDKN2A tumor suppressor gene encodes the p16INK4A protein, a highly specific endogenous inhibitor that binds to CDK4/6, physically preventing its association with Cyclin D.
- The Biological Deficit: In numerous aggressive malignancies—including melanomas, glioblastomas, and pancreatic ductal adenocarcinomas—the CDKN2A locus is either entirely deleted, epigenetically silenced via promoter hypermethylation, or inactivated via deleterious mutations.
- The Clinical Result: Deprived of the p16INK4A inhibitory constraint, the CDK4/6 complex operates unchecked, driving rapid and unrelenting cellular proliferation regardless of extracellular signaling.
Pathway Connectivity: The Rb-E2F Transcriptional Axis
To arrest tumor expansion, pharmacological interventions must disrupt the specific biochemical cascade the cancer relies upon. The core catalytic function of the Cyclin D-CDK4/6 complex involves the precise phosphorylation of the Retinoblastoma tumor suppressor protein (Rb).
- The Transcriptional Repressor (Rb): Within the cellular nucleus, the unphosphorylated Rb protein acts as a primary transcriptional repressor. It physically binds and sequesters the E2F family of transcription factors. While sequestered, E2F is transcriptionally inactive, preventing the cell from initiating DNA synthesis.
- Catalytic Phosphorylation: When the Cyclin D-CDK4/6 complex is activated by upstream mitogenic signals (or pathological amplification), it translocates to the nucleus and phosphorylates specific serine and threonine residues on the Rb protein.
- Conformational Shift and Release: This phosphorylation induces a massive conformational change in the Rb protein, disrupting its affinity for E2F. The Rb protein dissociates, releasing the E2F transcription factors.
- S-Phase Initiation: The liberated E2F proteins rapidly bind to promoter regions of target genes, initiating the transcription of machinery required for the S-phase (such as DNA polymerases and Cyclin E).
In CDK4/6-dysregulated tumors, the hyperactive kinase complex maintains Rb in a state of continuous hyperphosphorylation. The E2F factors are permanently liberated, resulting in aggressive, unchecked tumor replication.
Pharmacodynamics: Mechanism of CDK4/6 Inhibitors
For decades, advanced HR+/HER2- breast cancer was managed sequentially with endocrine therapies (e.g., aromatase inhibitors, selective estrogen receptor degraders), inevitably progressing to systemic cytotoxic chemotherapy upon the acquisition of endocrine resistance. The advent of highly selective, small-molecule CDK4/6 inhibitors (palbociclib, ribociclib, and abemaciclib) revolutionized this treatment paradigm.
ATP-Competitive Inhibition
These pharmacological agents are engineered to specifically target the catalytic domain of the CDK4 and CDK6 kinases. Like all kinases, CDK4/6 requires adenosine triphosphate (ATP) as a primary phosphate donor to execute the phosphorylation of the Rb protein.
- Intracellular Penetration: Following oral administration, these lipophilic small molecules enter the systemic circulation and penetrate the plasma membrane of the malignant cells.
- Cleft Occupation: The inhibitors function as reversible, ATP-competitive agents. They physically insert themselves into the highly conserved ATP-binding cleft of the CDK4 and CDK6 proteins.
- Catalytic Arrest: By occupying this specific molecular pocket, the drug sterically hinders ATP from binding. The kinase is rendered catalytically inert.
- Induction of Senescence: Deprived of CDK4/6 catalytic activity, the Rb protein remains in its unphosphorylated, active state. It permanently sequesters E2F. The tumor cell is arrested firmly at the G1 phase, preventing DNA replication. Prolonged G1 arrest frequently pushes the malignant cell into senescence (a state of permanent proliferative arrest) or triggers apoptotic pathways.

Clinical Efficacy and Landmark Trial Data
The integration of CDK4/6 inhibitors into the global standard of care is underpinned by rigorous, international Phase III clinical trials. These agents are currently the foundational first-line therapy for metastatic HR+/HER2- breast cancer, utilized synergistically with endocrine backbone therapies.
Progression-Free Survival (PFS)
When evaluating the efficacy of these agents across the PALOMA (palbociclib), MONALEESA (ribociclib), and MONARCH (abemaciclib) trial programs, the most immediate metric of success was Progression-Free Survival. Across multiple patient cohorts, the addition of a CDK4/6 inhibitor to standard endocrine therapy demonstrated a statistically significant and highly reproducible doubling of median PFS, effectively extending strict disease control by 10 to 14 months compared to endocrine therapy alone.
Overall Survival (OS) and Adjuvant Integration
Subsequent long-term data analysis has confirmed significant Overall Survival (OS) benefits, particularly noted in the MONALEESA trials evaluating ribociclib, demonstrating that targeted pathway inhibition profoundly alters the natural trajectory of the disease, delaying the necessity for cytotoxic chemotherapy and extending absolute patient lifespan.
Furthermore, the clinical success of these agents in the metastatic setting has catalyzed their transition into early-stage, curative-intent protocols. Abemaciclib and ribociclib have received regulatory approvals in the adjuvant setting for patients with high-risk, early-stage HR+/HER2- breast cancer, significantly reducing the statistical probability of systemic recurrence following primary surgical resection.
Target Specificity and Adverse Event Profiling
The primary clinical advantage of targeted kinase inhibitors lies in their target specificity, drastically altering the toxicity profile compared to traditional cytotoxic chemotherapy. Traditional chemotherapy induces widespread DNA damage in any rapidly dividing cell line, precipitating total alopecia, severe gastrointestinal mucositis, and profound immunosuppression.
CDK4/6 inhibitors selectively arrest the G1/S transition. Because the majority of healthy somatic tissue remains quiescent (in the G0 phase), it is largely bypassed by the pharmacological blockade. However, specific progenitor cells within the human body obligatorily cycle to maintain homeostasis.
On-Target Hematologic Toxicity (Neutropenia)
The hematopoietic stem cells within the bone marrow heavily rely on CDK6 to proliferate and generate neutrophil precursors. Consequently, the most prevalent, dose-limiting toxicity of CDK4/6 inhibition is neutropenia.
Crucially, CDK4/6-induced neutropenia is biologically distinct from chemotherapy-induced neutropenia. Chemotherapy causes the apoptotic death of bone marrow stem cells, necessitating prolonged recovery. Conversely, CDK4/6 inhibitors merely induce a temporary, reversible cell-cycle arrest in these progenitor cells. Upon holding the medication or initiating a standardized dose reduction, the bone marrow rapidly resumes proliferation, restoring absolute neutrophil counts without permanent architectural damage to the marrow.
Drug-Specific Toxicity Profiles
While class effects exist, slight molecular variances among the three approved inhibitors result in unique clinical toxicity profiles requiring specific monitoring:
- Abemaciclib: Exhibits greater continuous affinity for CDK4 over CDK6 and possesses a unique gastrointestinal toxicity profile, frequently inducing grade 1/2 diarrhea that requires proactive management with antidiarrheal agents (e.g., loperamide).
- Ribociclib: Associated with hepatic enzyme elevations and a dose-dependent prolongation of the QTc interval on electrocardiography, necessitating rigorous baseline and sequential ECG and hepatic monitoring during the first biological cycles.
Acquired Drug Resistance and Precision Sequencing
Malignant tumor biology is characterized by profound genomic plasticity. While CDK4/6 inhibitors are highly effective at inducing cellular senescence, the selective pressure exerted by the drug inevitably forces the tumor to adapt. The cessation of disease control indicates the acquisition of drug resistance. Resistance to CDK4/6 inhibitors is highly heterogeneous and manifests through specific, targetable genomic alterations.
1. Loss of the Retinoblastoma (Rb) Target
The ultimate pharmacological goal of CDK4/6 inhibition is the maintenance of unphosphorylated Rb. If the malignant cell acquires a somatic loss-of-function mutation in the RB1 gene, the target is effectively removed. Without the Rb protein to sequester E2F, the transcriptional factors flow freely, initiating the S-phase entirely independent of upstream CDK4/6 activity. RB1 mutations render continued CDK4/6 inhibition clinically futile.
2. Amplification of Compensatory Kinases (CDK2/Cyclin E)
Cellular networks are highly redundant. In the presence of a sustained CDK4/6 blockade, the tumor may heavily amplify the transcription of Cyclin E and its catalytic partner, CDK2. The active Cyclin E-CDK2 complex functions downstream of CDK4/6 and possesses the catalytic capacity to independently phosphorylate Rb. This creates a biological bypass, restoring E2F release and re-initiating the cell cycle.
3. Upstream Pathway Hyperactivation
Tumors frequently acquire secondary activating mutations in parallel oncogenic pathways, most notably the PI3K/AKT/mTOR signaling cascade (e.g., PIK3CA or AKT1 mutations) or the FGFR (Fibroblast Growth Factor Receptor) pathway. These hyperactive networks generate massive intracellular survival signals that functionally overwhelm the G1 arrest induced by CDK4/6 inhibition.

Clinical Solutions and Next-Generation Interventions
The identification of acquired resistance necessitates immediate clinical reassessment. Contemporary oncological protocols utilize comprehensive liquid biopsies (ctDNA sequencing) to non-invasively map the tumor’s newly adapted genomic landscape at the point of progression.
Understanding the precise mechanism of resistance dictates subsequent therapeutic sequencing. If NGS reveals an acquired PIK3CA or AKT1 mutation, targeted inhibitors (such as alpelisib or capivasertib) can be strategically deployed. Furthermore, extensive clinical trial frameworks are actively evaluating next-generation pharmacological agents, including highly selective CDK2 inhibitors and targeted protein degraders, designed specifically to dismantle the bypass mechanisms and restore strict disease control.
Through the continuous integration of molecular profiling and targeted pharmacology, the clinical armamentarium adapts to the evolutionary dynamics of the malignancy, ensuring that treatment remains rigorously precise, scientifically grounded, and profoundly effective.
What exactly is a CDK4/6 inhibitor?
CDK4/6 inhibitors are targeted oral medications (like palbociclib, ribociclib, and abemaciclib) that block specific proteins driving cancer cell division. By binding to the CDK4 and CDK6 kinases, these drugs trap the cancer cell in a resting state (G1 phase arrest). This halts DNA replication and stops the tumor from growing, forcing the malignant cells into permanent senescence or programmed death.
Are CDK4/6 inhibitors considered traditional chemotherapy?
What specific cancers are treated with CDK4/6 inhibitors?
Do CDK4/6 inhibitors cause severe hair loss (alopecia)?
Why do these targeted therapies cause neutropenia (low white blood cells)?
How long do CDK4/6 inhibitors keep the cancer controlled?
What is acquired resistance to CDK4/6 inhibitors?
Can CDK4/6 inhibitors be taken without hormone therapy?
Are there specific dietary restrictions while taking these inhibitors?
Disclaimer: The information provided in this comprehensive article is strictly for educational and informational purposes. It is completely independent and objective. This content is not a substitute for professional medical advice, formal diagnosis, or specialized clinical treatment. Always consult with your board-certified oncologist, endocrinologist, or designated healthcare provider regarding your specific medical condition, comprehensive genomic testing results, and customized treatment protocol. Medically Reviewed by Dr. Salma Elreedy, MD (Clinical Oncology), Sphinx Cure Oncology Center.
Educational References
The biological mechanisms, genetic data, and clinical trial statistics detailed in this comprehensive guide are grounded in rigorous, peer-reviewed science published by authoritative medical institutions. For primary source data and detailed clinical guidelines, consult the following resources:
The National Center for Biotechnology Information (NCBI) / PubMed
The American Society of Clinical Oncology (ASCO)